Paraganglioma
Overview
Paragangliomas are masses derived from the paraganglia, a group of non-neuronal cells that are associated with the sympathetic and parasympathetic nervous system.
History
These tumors historically were called "glomus tumors" when present in the head and neck. This term has fallen out of use due to potential confusion with other structures with similar names, such as glomus bodies.
Pathophysiology
Relevant Anatomy
The term "paraganglia" refers to a group of non-neuronal neuroendocrine cells derived embryologically from neural crest cells. There are paraganglia associated with the sympathetic nervous system comprised of chromaffin cells, and those associated with the parasympathetic nervous system comprised of glomus cells. The adrenal medulla is the largest collection of Chromaffin cells in the body. The paraganglia of the head and neck region are associated with the glomus cells of the parasympathetic nervous system.
The paraganglia are highly vascularized for the purposes of chemoreceptor sensitivity. Sympathetic paraganglia act as endocrine organs with systemic catecholamine release (such as the adrenal medulla, organ of Zuckerkandl, etc.). These paraganglia are a major source of catecholamines in early embryogenesis (assuming much function of the adrenal medulla). Parasympathetic paraganglia predominantly have more local effects on nerve endings, such as in the carotid body.
-

Target end-organs of the sympathetic nervous system.
-

Target end-organs of the parasympathetic nervous system.


Disease Etiology
Paragangliomas are a result of unregulated growth of paragangliar tissue. A majority of these masses arise from the carotid body just superior to the carotid bifurcation. Paragangliomas that arise from the area around the temporal bone are termed jugulotympanic paragangliomas. The term glomus tympanicum historically describes paragangliomas that arise from Jacobson's nerve (the tympanic branch of CN XI) - by definition these arise on the cochlea promontory and are confined to the middle ear and mastoid cavity. The term glomus jugulare was previously used to describe paragangliomas that arise along Jacobson's nerve or Arnold's nerve (the auricular branch of CN X), and involve the jugular bulb or skull base. These two terms have fallen out of favor and are more commonly described now collectively as "jugulotympanic" paragangliomas.
In rare instances these masses have been documented to metastasize, in what is termed a "malignant paraganglioma". Typical metastases are to the cervical lymph nodes but distant metastases to the bone, liver, and lung have also been described. Of note, syndromic paragangliomas are less often malignant compared to sporadic disease.
Epidemiology
The incidence of head and neck paragangliomas has been reported as 1 in 30,000 to 1 in 100,000. Approximately 10% of patients with head and neck paragangliomas will have multiple masses. As many as 25-35% of cases have been linked to hereditary conditions, typically Familial Paraganglioma Syndrome (see Genetics section below). There is an equal distribution of carotid paragangliomas in men and women, but jugulotympanic paragangliomas are six times more likely to be in women.
Genetics
There are several genetic syndromes associated with paragangliomas of the head and neck.[1]
| Syndrome | Gene | Locus | Inheritance | Additional Findings |
|---|---|---|---|---|
| Paraganglioma Syndrome Type 1 (PGL1) | SDHD | 11q23.1 | AD, Maternal Imprinting | Pituitary adenoma, GIST, RCC |
| Paraganglioma Syndrome Type 2 (PGL2) | SDHAF2 | 11q13.1 | AD, Maternal Imprinting | Pituitary adenoma, GIST, RCC |
| Paraganglioma Syndrome Type 3 (PGL3) | SDHC | 1q23.3 | AD | Pituitary adenoma, GIST, RCC |
| Paraganglioma Syndrome Type 4 (PGL4) | SDHB | 1p36.13 | AD | Pituitary adenoma, GIST, RCC |
| Paraganglioma Syndrome Type 5 (PGL5) | SDHA | 5p15.33 | AD | Pituitary adenoma, GIST, RCC |
| von Hippel-Lindau Syndrome | VHL | 3p25.3 | AD | Hemangioblastoma, retinal angioma, clear cell RCC, pancreatic neuroendocrine tumors, endolymphatic sac tumors |
| Multiple Endocrine Neoplasia type 2 (MEN-2) | RET | 10q11.21 | AD | Medullary thyroid carcinoma, Hyperparathyroidism |
| Neurofibromatosis Type 1 (NF1) | NF1 | 17q11.2 | AD | Neurofibromas, cafe-au-lait spots, cognitive deficits, Lisch nodules |
Histology
There are two predominant cell types seen on histologic sections of paragangliomas. Type I cells, also known as Glomus Cells, are comprised of secretory cells and make up a majority of the bulk of paragangliomas. Type I cells will stain positive for neuroendocrine markers such as chromogranin, synaptophysin, neuron-specific enolase (NSE), and CD56. Type II cells, also known as Sustentacular Cells, is comprised of Schwann-like satellite cells. Type II cells stain positive for S100 and occasionally glial fibrillary acidic protein (GFAP). Paragangliomas will arrange themselves in the same histologic morphology as normal paraganglia tissue, in what is described as the Zellballen configuration - an arrangement of clusters of Type I cells in a surrounding stroma of Type II cells.[2]
-
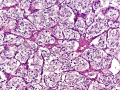
Paraganglioma section at 20x demonstrating the Zellballen configuration.
-
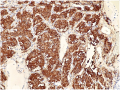
Synaptophysin staining highlighting the Type I cells.
-
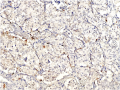
S100 staining highlighting the Type II cells.



Diagnosis
Patient History
Jugulotympanic paragangliomas classically present in the patient's fourth or fifth decade of life with progressive hearing loss and pulsatile tinnitus.[3] This is often a slowly progressive, insidious loss of hearing with mass identification months to years after symptom onset. They may also have bloody otorrhea or vertigo. Depending on the extent of the disease at the time, the patient may have erosion into the adjacent skull base foramina and associated cranial neuropathy.
Carotid body paragangliomas typically present as a slow-growing painless lateral neck mass.[4] In advanced disease, patients may have hoarseness from CN X involvement or decreased tongue mobility from CN XII involvement.
Physical Examination

Patients with a jugulotympanic paraganglioma may have a pulsatile red mass seen behind the medial tympanic membrane (known as the "Rising Sun Sign"). Due to the hypervascularity of these masses, they may blanch on pneumatic otoscopy (known as "Brown's sign").
Patients with carotid body paragangliomas will present with a painless mass in the lateral neck. Due to the attachment to the carotid vessels, this will be more mobile in a lateral direction than a cranial/caudal direction (known as "Fontaine's sign"). These masses are often pulsatile and will have an audible bruit on auscultation.
Patients with suspected paragangliomas may have cranial nerve deficits and should undergo a rigorous cranial nerve exam.
Laboratory Tests
Laboratory workup of paragangliomas should focus identifying whether the mass is a secretory paraganglioma by assessing for the presence of metanephrines and/or their metabolites:
- Urine metanephrines
- Urine vanillylmandelic acid (VMA)
- Urine homovanillic acid (HVA)
- Plasma metanephrines
- Plasma vanillylmandelic acid (VMA)
- Plasma homovanillic acid (HVA)
These are typically expensive tests that will need to be sent out. They may be bundled or separate orders based on your institution. This is important information for anesthesia purposes if you are planning a surgical resection. It is important to note that these masses should not be biopsied for tissue diagnosis if you suspect paraganglioma. They are extremely vascular and present a high risk for hemorrhage.
Imaging
Most paragangliomas of the head and neck are initially characterized on CT scan during the workup of a new neck mass. Paragangliomas will typically avidly enhance with contrast and have a delayed washout due to their rich capillary network. When the paraganglioma approaches the skull base, you may also see erosion of adjacent bone and involvement/compression of skull base foramina.
On MRI, paragangliomas will be T1 hypointense and T2 hyperintense, with a classic "salt and pepper" appearance on T1 imaging. This is due to the flow voids in the vascular network, with the "salt" describing the bulk of the mass and the "pepper" being the dark spots of the flow voids.[5]
Angiography will demonstrate a classic "Lyre sign", in which the internal and external carotid arteries are splayed apart from the mass effect between them.
-

Carotid body tumor angiography with the internal and external carotid artery separated by mass effect.
-

A Lyre, for reference.


The extent of the mass, if in the carotid body, is often described by the Shamblin classification.[6]
| Type I | Type II | Type III |
|---|---|---|
 |
 |
 |
| Relatively small with minimal attachment to the carotid vessels | Somewhat larger with more extensive attachment to the carotid vessels | Large mass with complete encasement of the carotid vessels |
Differential Diagnosis
Patients with paragangliomas, particularly in the jugulotympanic distribution, can present with the various eponymous jugular foramen syndromes:
| Syndrome | CN IX | CN X | CN XI | CN XII | Sympathetics |
|---|---|---|---|---|---|
| Vernet Syndrome | ✔ | ✔ | ✔ | ||
| Collet-Sicard Syndrome | ✔ | ✔ | ✔ | ✔ | |
| Villaret Syndrome | ✔ | ✔ | ✔ | ✔ | ✔ |
| Tapia Syndrome | ✔ | ± | ✔ | ± | |
| Jackson Syndrome | ✔ | ✔ | ✔ | ||
| Schmidt Syndrome | ✔ | ✔ |
There are several paraganglioma-associated syndromes that should be considered if the patient has any of the associated characteristic findings of each:
- von Hippel-Lindau (VHL) Syndrome
- Multiple Endocrine Neoplasia type 2 (MEN-2)
- Neurofibromatosis type 1 (NF1)
- Succinate Dehydrogenase-associated paraganglioma syndromes
In addition, the following diagnoses should be considered for patients with new neck masses:
- Schwannoma
- Arterio-Venous Malformation
- Metastatic Malignancy
Management
Medical Management
Not all patients require surgical intervention. For patients that are not interested in surgery, or are poor surgical candidates, observation is an acceptable option. These are slow-growing tumors at a typical rate of approximately 1 to 2 mm per year, with some patients exhibiting no growth at all.[7][8] Many providers will opt for annual interval imaging studies to track the growth rate and assess whether the patient is developing new symptoms. Radiation therapy is also an option to limit the growth rate of head and neck paragangliomas.[9][10]
Surgical Management
Surgical excision of paragangliomas can be challenging. The hypervascularity of the mass can result in significant intraoperative blood loss and obscuring of surgical planes. The close proximity of critical vascular and neural structures can also result in significant postoperative morbidity. Preoperative angiography with embolization 24 to 48 hours before surgery may decrease intraoperative bleeding and facilitate excision.
For carotid body paragangliomas, patients with a Shamblin Type I or Type II tumor are more amenable to surgical resection.[11] Patients with a Shamblin Type III carotid body mass may require arterial bypass and resection, for which vascular surgery should be involved.
Traditionally, jugulotympanic paragangliomas were managed surgically with an infratemporal fossa approach. This is associated with significant postoperative morbidity including hearing loss and facial paralysis. Conservative measures are more favorable where possible for these patients. If there is intracranial extension of the mass, this often requires a combined approach with surgical removal of the middle ear and mastoid components and intracranial radiation.
Outcomes
Complications
Carotid body paragangliomas run the risk of injury to the internal carotid, external carotid, CN X, CN XII, superior laryngeal nerve, and sympathetic chain. Due to the intimate relationship with the carotid vessels these patients are at risk of intraoperative/postoperative ischemic stroke and death, particularly among those with a Shamblin Type III mass.[12]
Prognosis
For patients that elect to observe these masses, there is typically minimal progression in symptoms on a yearly basis. In many cases, they will continue to grow slowly. In one study of 16 jugulotympanic paragangliomas managed by primary observation, primarily in elderly patients, there was minimal progressive compressive cranial neuropathy that patients typically compensated for well.[13] In the absence of concerns for malignancy or intracranial involvement, these masses are typically safe to observe.
References
- ↑ Tsirlin A, Oo Y, Sharma R, Kansara A, Gliwa A, Banerji MA. Pheochromocytoma: a review. Maturitas. 2014 Mar 1;77(3):229-38.
- ↑ Pernick N. Middle ear paraganglioma. PathologyOutlines.com website. https://www.pathologyoutlines.com/topic/earjugulotympanicparaganglioma.html. Accessed September 15th, 2024.
- ↑ Carlson ML, Sweeney AD, Wanna GB, Netterville JL, Haynes DS. Natural history of glomus jugulare: a review of 16 tumors managed with primary observation. Otolaryngology--Head and Neck Surgery. 2015 Jan;152(1):98-105.
- ↑ Moore MG, Netterville JL, Mendenhall WM, Isaacson B, Nussenbaum B. Head and neck paragangliomas: an update on evaluation and management. Otolaryngology--Head and Neck Surgery. 2016 Apr;154(4):597-605.
- ↑ Gaillard F, Campos A, Hartung M, et al. Paraganglioma. Reference article, Radiopaedia.org (Accessed on 11 Sep 2024) https://doi.org/10.53347/rID-1843
- ↑ Shamblin WR, ReMine WH, Sheps SG, Harrison Jr EG. Carotid body tumor (chemodectoma): clinicopathologic analysis of ninety cases. The American Journal of Surgery. 1971 Dec 1;122(6):732-9.
- ↑ Langerman A, Athavale SM, Rangarajan SV, Sinard RJ, Netterville JL. Natural history of cervical paragangliomas: outcomes of observation of 43 patients. Archives of Otolaryngology–Head & Neck Surgery. 2012 Apr 1;138(4):341-5.
- ↑ Carlson ML, Sweeney AD, Wanna GB, Netterville JL, Haynes DS. Natural history of glomus jugulare: a review of 16 tumors managed with primary observation. Otolaryngology--Head and Neck Surgery. 2015 Jan;152(1):98-105.
- ↑ Hinerman RW, Amdur RJ, Morris CG, Kirwan J, Mendenhall WM. Definitive radiotherapy in the management of paragangliomas arising in the head and neck: a 35‐year experience. Head & Neck: Journal for the Sciences and Specialties of the Head and Neck. 2008 Nov;30(11):1431-8.
- ↑ Gilbo P, Morris CG, Amdur RJ, Werning JW, Dziegielewski PT, Kirwan J, Mendenhall WM. Radiotherapy for benign head and neck paragangliomas: a 45‐year experience. Cancer. 2014 Dec 1;120(23):3738-43.
- ↑ Lim JY, Kim J, Kim SH, Lee S, Lim YC, Kim JW, Choi EC. Surgical treatment of carotid body paragangliomas: outcomes and complications according to the shamblin classification. Clinical and experimental otorhinolaryngology. 2010 Jun 30;3(2):91-5.
- ↑ Lim JY, Kim J, Kim SH, Lee S, Lim YC, Kim JW, Choi EC. Surgical treatment of carotid body paragangliomas: outcomes and complications according to the shamblin classification. Clinical and experimental otorhinolaryngology. 2010 Jun 30;3(2):91-5.
- ↑ Carlson ML, Sweeney AD, Wanna GB, Netterville JL, Haynes DS. Natural history of glomus jugulare: a review of 16 tumors managed with primary observation. Otolaryngology--Head and Neck Surgery. 2015 Jan;152(1):98-105.